Over the past century, the development of materials science has increasingly relied on the precise determination of atomic arrangements—that is, the crystal structure and its properties. To this end, X-ray diffraction (XRD) is commonly applied, with the sine qua non being the availability of a single crystal or monocrystal. However, this is not always feasible, especially with atomic clusters of nanometric size (smaller than 1000 Å), known as the nanostructure problem. In such cases, powder X-ray diffraction (PXRD) patterns are degraded due to peak broadening, intensity loss, and Bragg peak overlap.
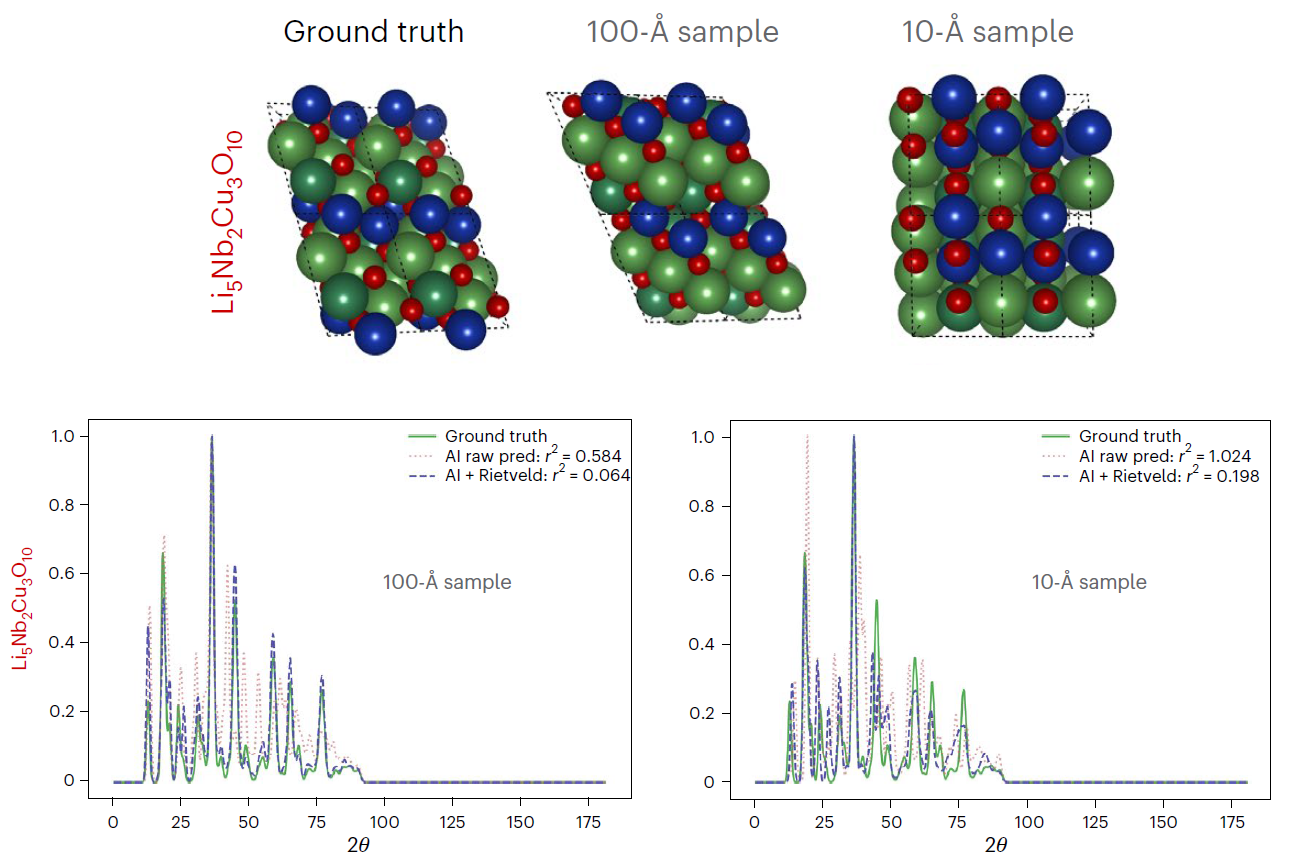
Researchers from the United States and Germany have proposed a procedure that uses a generative machine learning model* based on diffusion processes, trained on 45,229 known structures. The model, called PXRDnet, conditioned solely on the compound's chemical formula, can solve simulated nanocrystals up to 10 Å in 200 materials with various symmetries and complexities, including all seven crystal systems.
PXRDnet correctly identifies structural candidates in 4 out of 5 cases, with an average error of just 7% in the Rietveld refinement factor R. Moreover, it is capable of resolving structures from noisy experimentally obtained diffraction patterns.
The authors argue that this data-driven, theoretically bootstrapped approach opens new avenues for determining previously unsolved nanomaterial structures. However, the model has limitations: it requires prior knowledge of the chemical formula and is restricted to structures with fewer than 20 atoms per unit cell.
*The term “generative” refers to a class of statistical models as opposed to discriminative models. Generative models can generate new data instances, while discriminative models distinguish between different types of data instances.
The work was published by Nature Materials




No hay comentarios:
Publicar un comentario